Learning Objectives
- Define population structure
- Go through examples of manifestation of population strucutre
- Learn methods to correct for population structure
- genomic control
- principal component analysis
Material for the class
- Download notes on population structure from here
Prerequisites for running the analysis below
- plink installed in
~/bin/ - Rstudio is installed
- tidyverse package installed
- devtools package installed
download data from here TODO box link here
if(!file.exists(glue::glue("~/Downloads/analysis_population_structure.tgz")))
{
system(glue::glue("wget -O ~/Downloads/analysis_population_structure.tgz https://uchicago.box.com/shared/static/zv1jyevq01mt130ishx25sgb1agdu8lj.tgz"))
## tar -xf file_name.tar.gz --directory /target/directory
system(glue::glue("tar xvf ~/Downloads/analysis_population_structure.tgz --directory ~/Downloads/"))
}
work.dir ="~/Downloads/analysis_population_structure/"
system(glue::glue("tree {work.dir}"))library(tidyverse)
## load qqunif function
devtools::source_gist("38431b74c6c0bf90c12f")Test Hardy Weinberg equilibrium with population structure
What’s the population composition?
popinfo = read_tsv(paste0(work.dir,"relationships_w_pops_051208.txt"))##
## ── Column specification ────────────────────────────────────────────────────────
## cols(
## FID = col_character(),
## IID = col_character(),
## dad = col_character(),
## mom = col_character(),
## sex = col_double(),
## pheno = col_double(),
## population = col_character()
## )popinfo %>% count(population)## # A tibble: 11 x 2
## population n
## * <chr> <int>
## 1 ASW 90
## 2 CEU 180
## 3 CHB 90
## 4 CHD 100
## 5 GIH 100
## 6 JPT 91
## 7 LWK 100
## 8 MEX 90
## 9 MKK 180
## 10 TSI 100
## 11 YRI 180samdata = read_tsv(paste0(work.dir,"phase3_corrected.psam"),guess_max = 2500) ##
## ── Column specification ────────────────────────────────────────────────────────
## cols(
## `#IID` = col_character(),
## PAT = col_character(),
## MAT = col_character(),
## SEX = col_double(),
## SuperPop = col_character(),
## Population = col_character()
## )superpop = samdata %>% select(SuperPop,Population) %>% unique()
superpop = rbind(superpop, data.frame(SuperPop=c("EAS","HIS","AFR"),Population=c("CHD","MEX","MKK")))Effect of population structure in Hardy Weinberg equilibrium
## what happens if we calculate HWE with this mixed population?
if(!file.exists(glue::glue("{work.dir}output/allhwe.hwe")))
system(glue::glue("~/bin/plink --bfile {work.dir}hapmapch22 --hardy --out {work.dir}output/allhwe"))
allhwe = read.table(glue::glue("{work.dir}output/allhwe.hwe"),header=TRUE,as.is=TRUE)
hist(allhwe$P)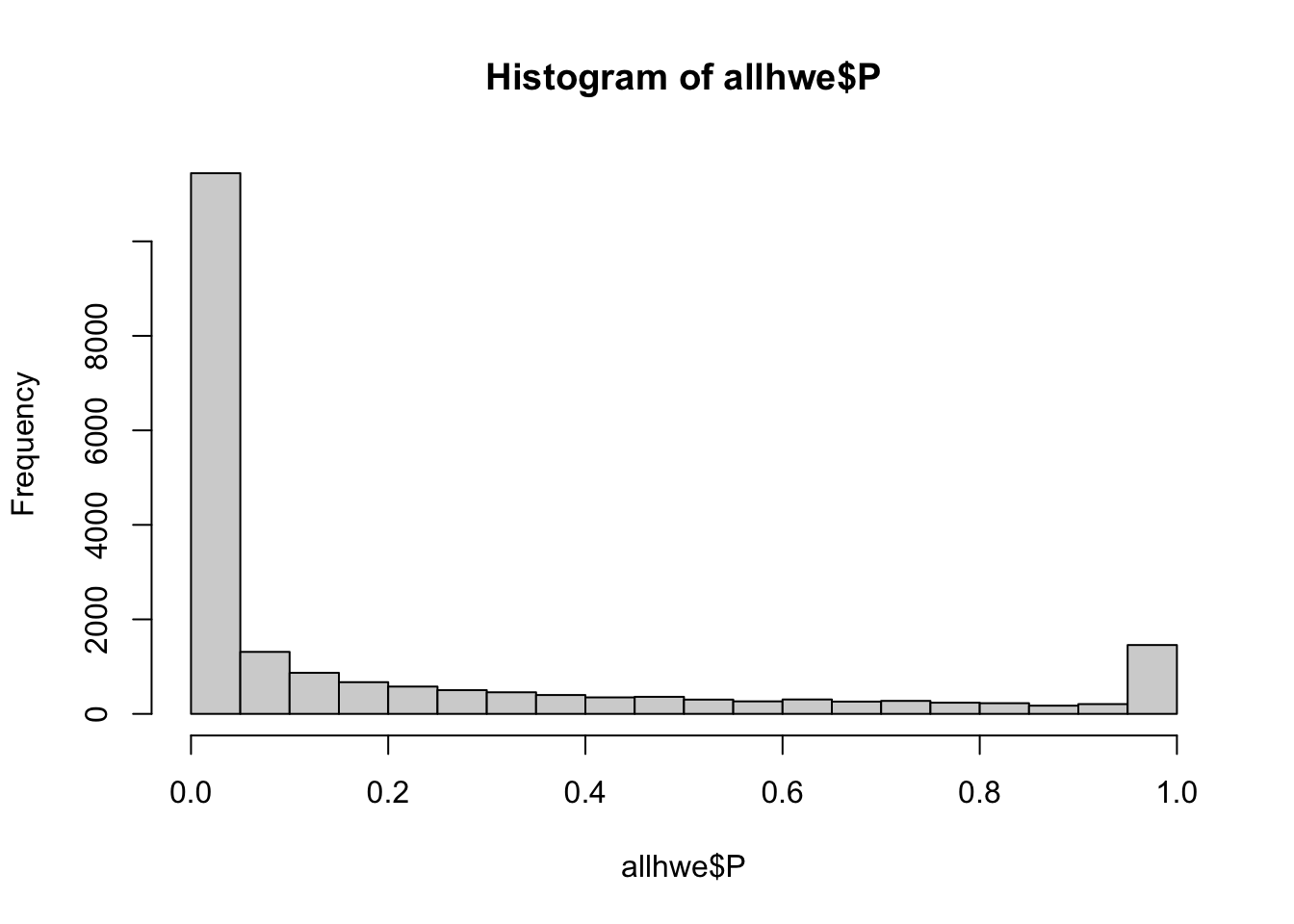
qqunif(allhwe$P,main='HWE HapMap3 All Pop')## Warning in qqunif(allhwe$P, main = "HWE HapMap3 All Pop"): thresholding p to
## 1e-30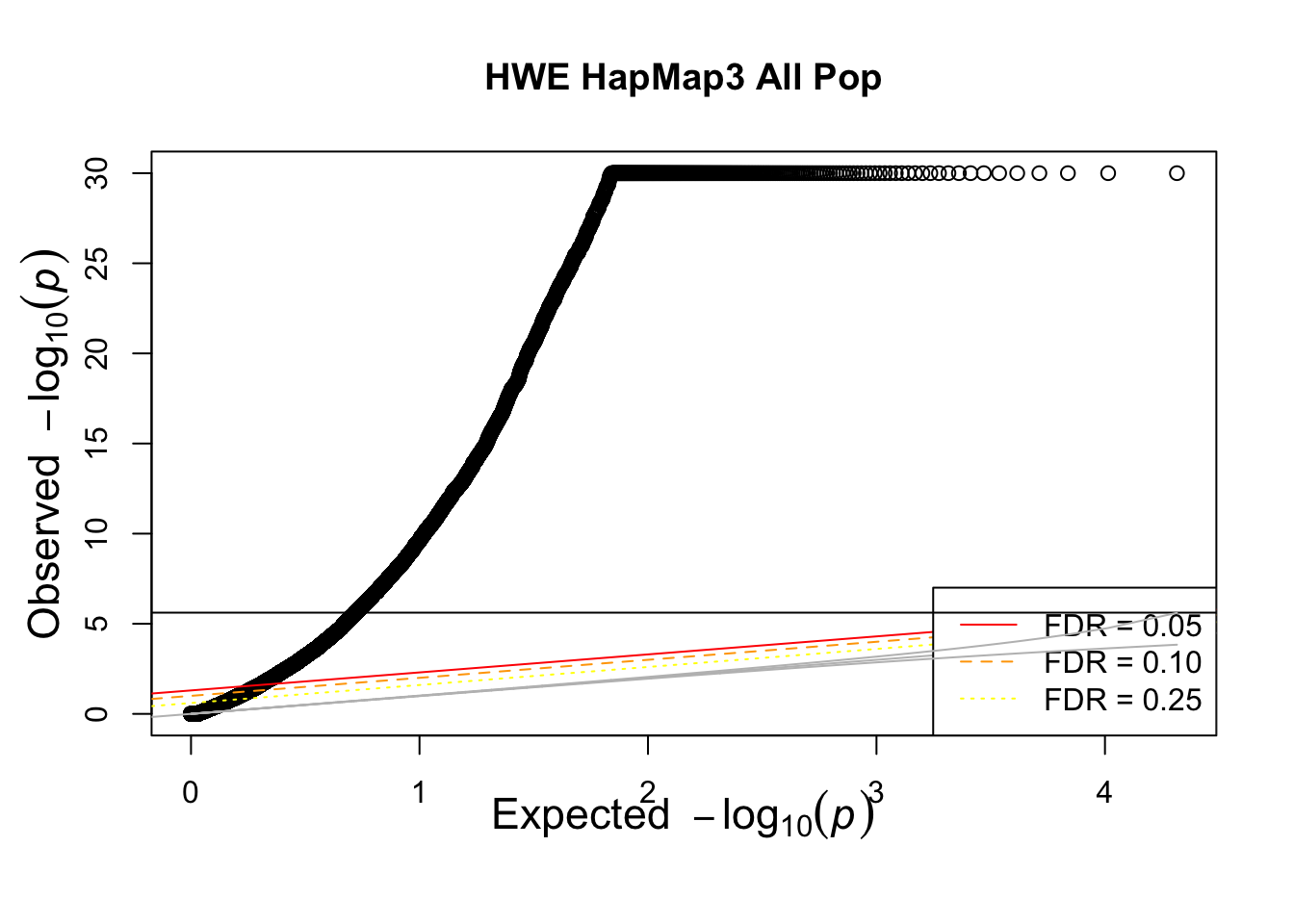
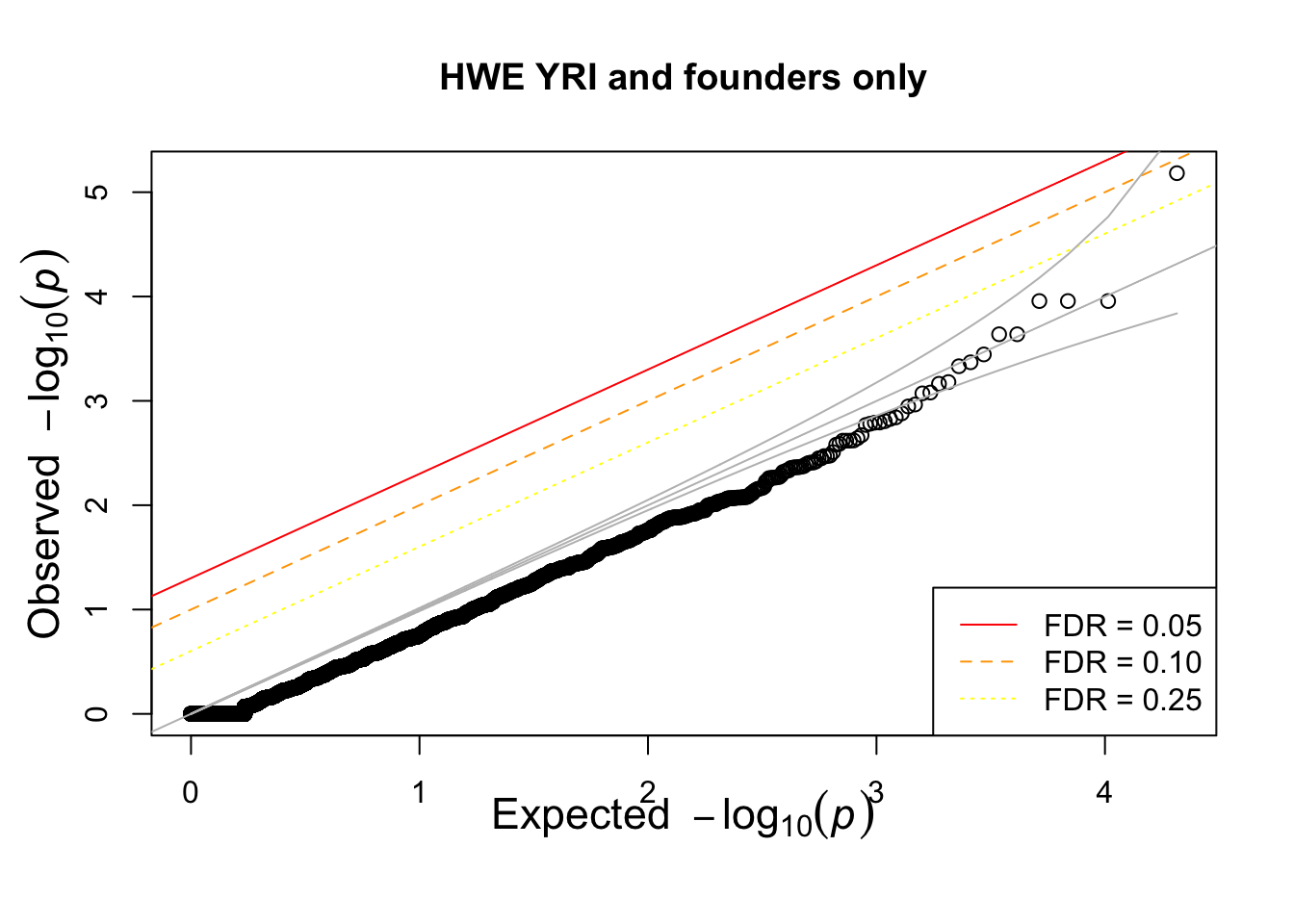
What if we calculate with single population?
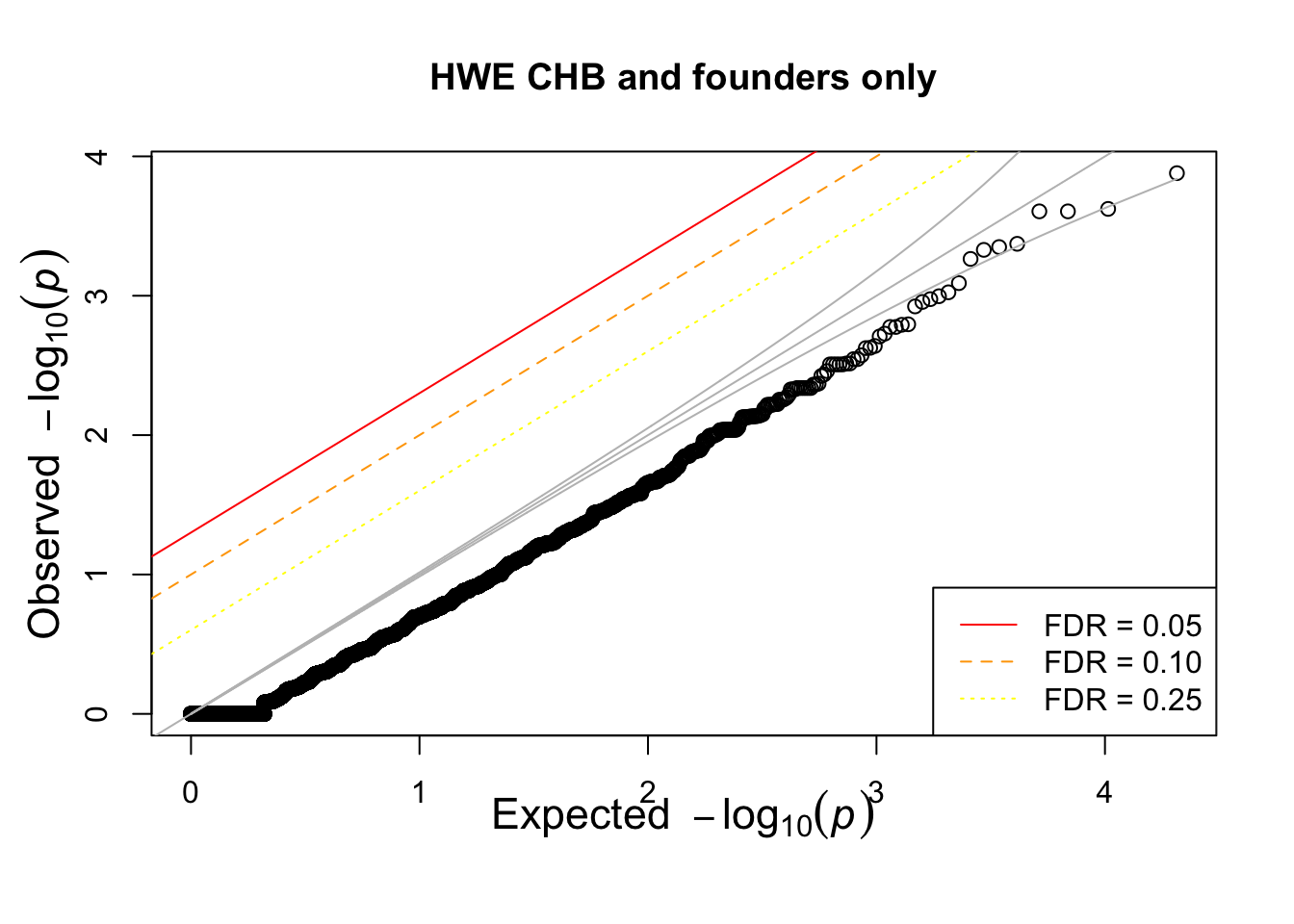
pop = "CHB"
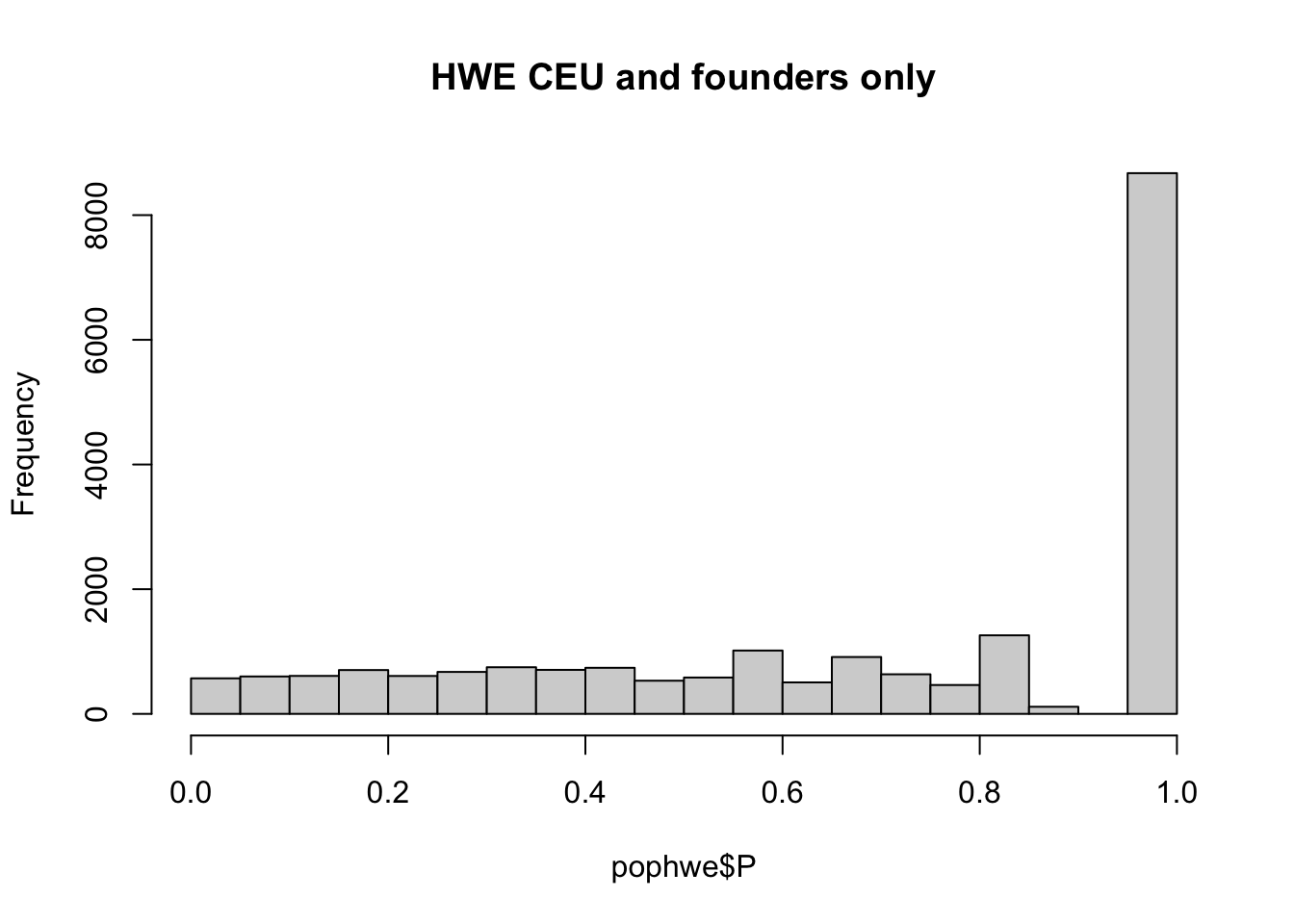
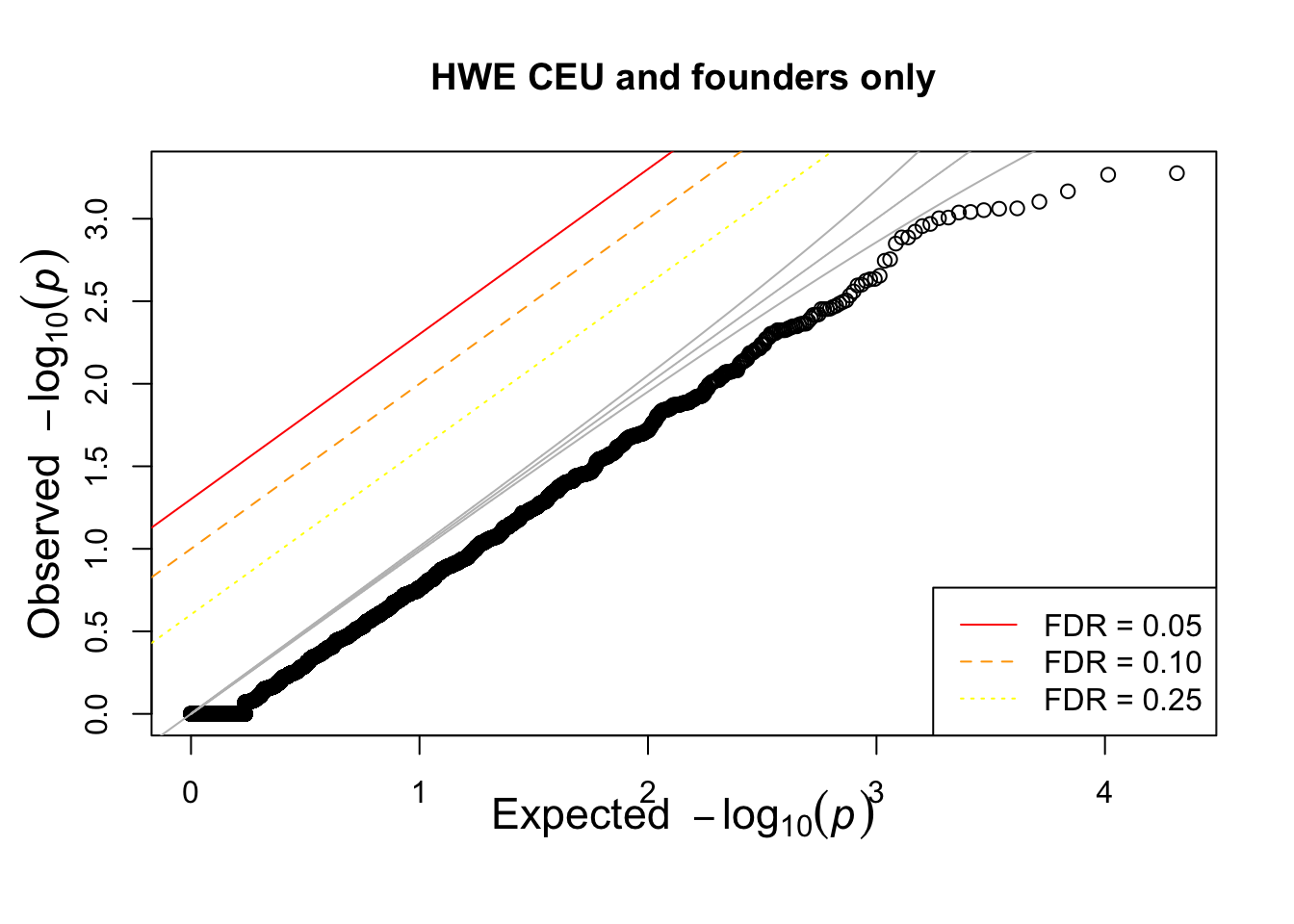
pop = "CEU"
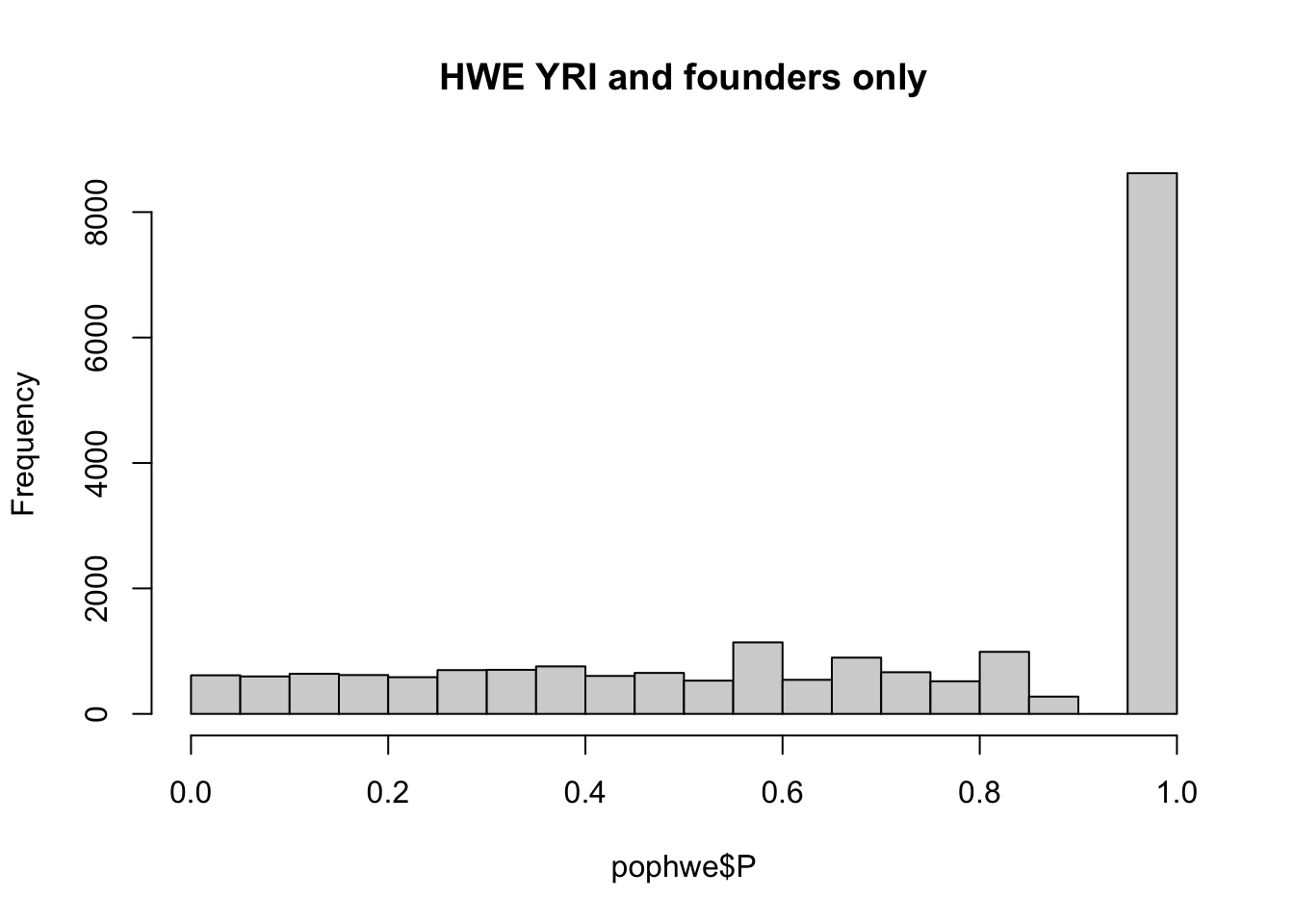
pop = "YRI"
for(pop in c("CHB","CEU","YRI"))
{
## what if we calculate with single population?
popinfo %>% filter(population==pop) %>%
write_tsv(path=glue::glue("{work.dir}{pop}.fam") )
if(!file.exists(glue::glue("{work.dir}output/hwe-{pop}.hwe")))
system(glue::glue("~/bin/plink --bfile {work.dir}hapmapch22 --hardy --keep {work.dir}{pop}.fam --out {work.dir}output/hwe-{pop}"))
pophwe = read.table(glue::glue("{work.dir}output/hwe-{pop}.hwe"),header=TRUE,as.is=TRUE)
hist(pophwe$P,main=glue::glue("HWE {pop} and founders only"))
qqunif(pophwe$P,main=glue::glue("HWE {pop} and founders only"))
}## Warning: The `path` argument of `write_tsv()` is deprecated as of readr 1.4.0.
## Please use the `file` argument instead.
## This warning is displayed once every 8 hours.
## Call `lifecycle::last_warnings()` to see where this warning was generated.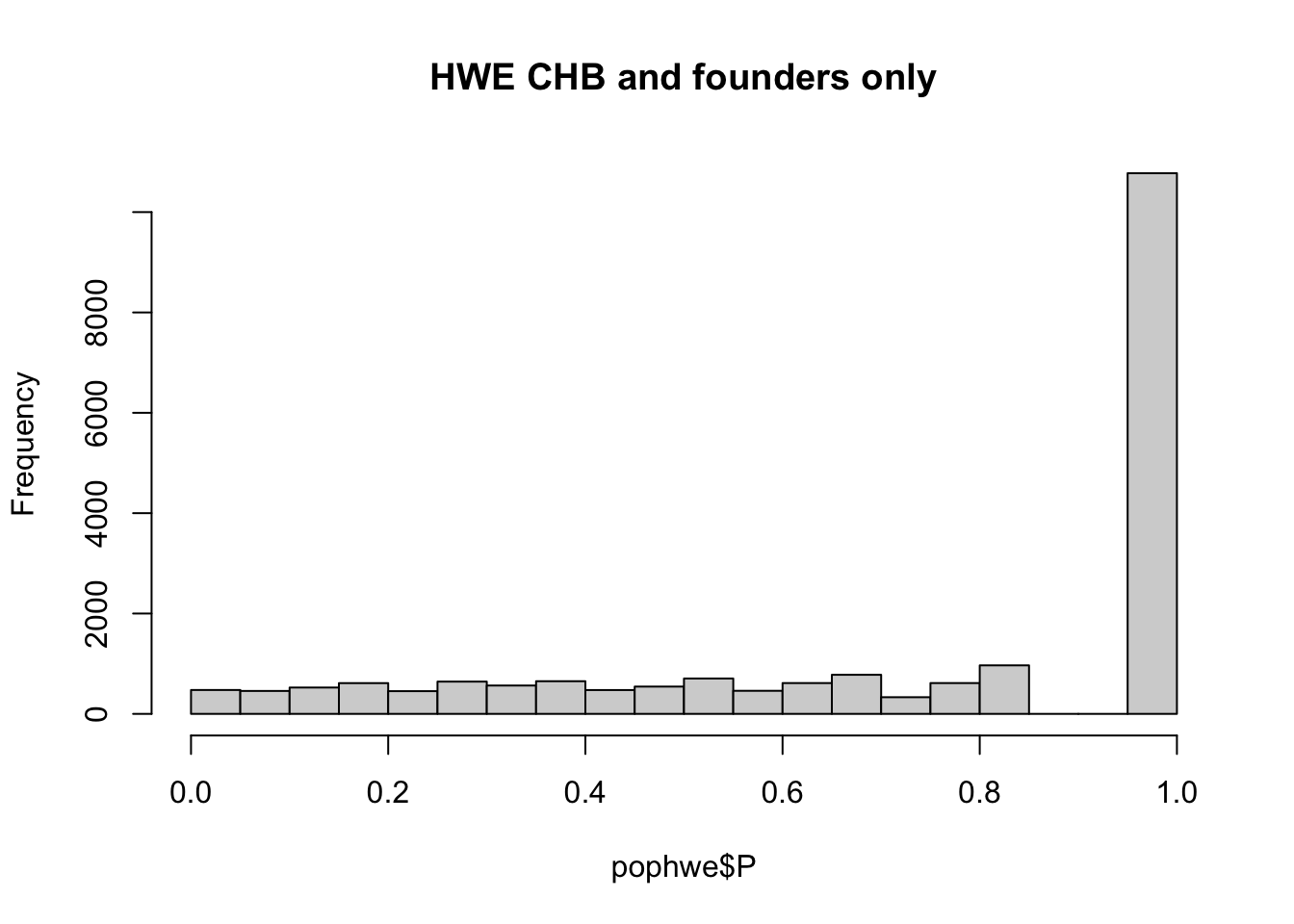
Effect of population stratification on GWAS
GWAS on a growth phenotype in HapMap samples
## read igrowth
igrowth = read_tsv("https://raw.githubusercontent.com/hakyimlab/igrowth/master/rawgrowth.txt")##
## ── Column specification ────────────────────────────────────────────────────────
## cols(
## IID = col_character(),
## sex = col_double(),
## pop = col_character(),
## experim = col_double(),
## meas.by = col_double(),
## serum = col_character(),
## growth = col_double()
## )## add FID to igrowth file
igrowth = popinfo %>% select(-pheno) %>% inner_join(igrowth %>% select(IID,growth), by=c("IID"="IID"))
write_tsv(igrowth,path=glue::glue("{work.dir}igrowth.pheno"))
igrowth %>% ggplot(aes(population,growth)) + geom_violin(aes(fill=population)) + geom_boxplot(width=0.2,col='black',fill='gray',alpha=.8) + theme_bw(base_size = 15)## Warning: Removed 130 rows containing non-finite values (stat_ydensity).## Warning: Removed 130 rows containing non-finite values (stat_boxplot).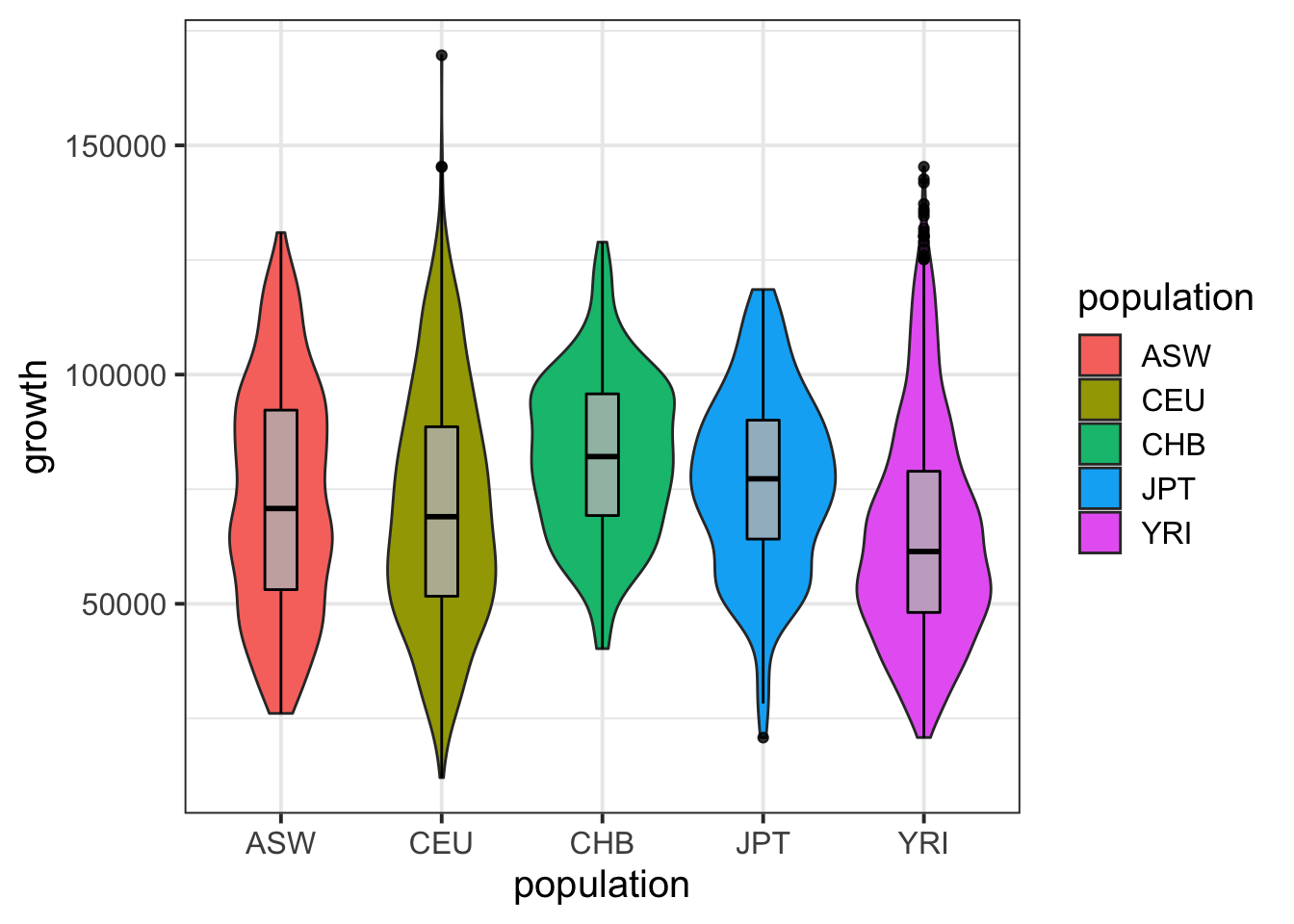
summary( lm(growth~population,data=igrowth) )##
## Call:
## lm(formula = growth ~ population, data = igrowth)
##
## Residuals:
## Min 1Q Median 3Q Max
## -58821 -18093 -2242 15896 98760
##
## Coefficients:
## Estimate Std. Error t value Pr(>|t|)
## (Intercept) 73080.8 938.2 77.894 < 2e-16 ***
## populationCEU -2190.1 1175.4 -1.863 0.0625 .
## populationCHB 9053.1 2043.9 4.429 9.73e-06 ***
## populationJPT 3476.8 2034.8 1.709 0.0876 .
## populationYRI -7985.2 1137.2 -7.022 2.61e-12 ***
## ---
## Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
##
## Residual standard error: 24160 on 3591 degrees of freedom
## (130 observations deleted due to missingness)
## Multiple R-squared: 0.0345, Adjusted R-squared: 0.03342
## F-statistic: 32.08 on 4 and 3591 DF, p-value: < 2.2e-16## run plink linear regression only if it hasn't been run already
if(!file.exists(glue::glue("{work.dir}output/igrowth.assoc.linear")))
system(glue::glue("~/bin/plink --bfile {work.dir}hapmapch22 --linear --pheno {work.dir}igrowth.pheno --pheno-name growth --maf 0.05 --out {work.dir}output/igrowth"))
igrowth.assoc = read.table(glue::glue("{work.dir}output/igrowth.assoc.linear"),header=T,as.is=T)
hist(igrowth.assoc$P)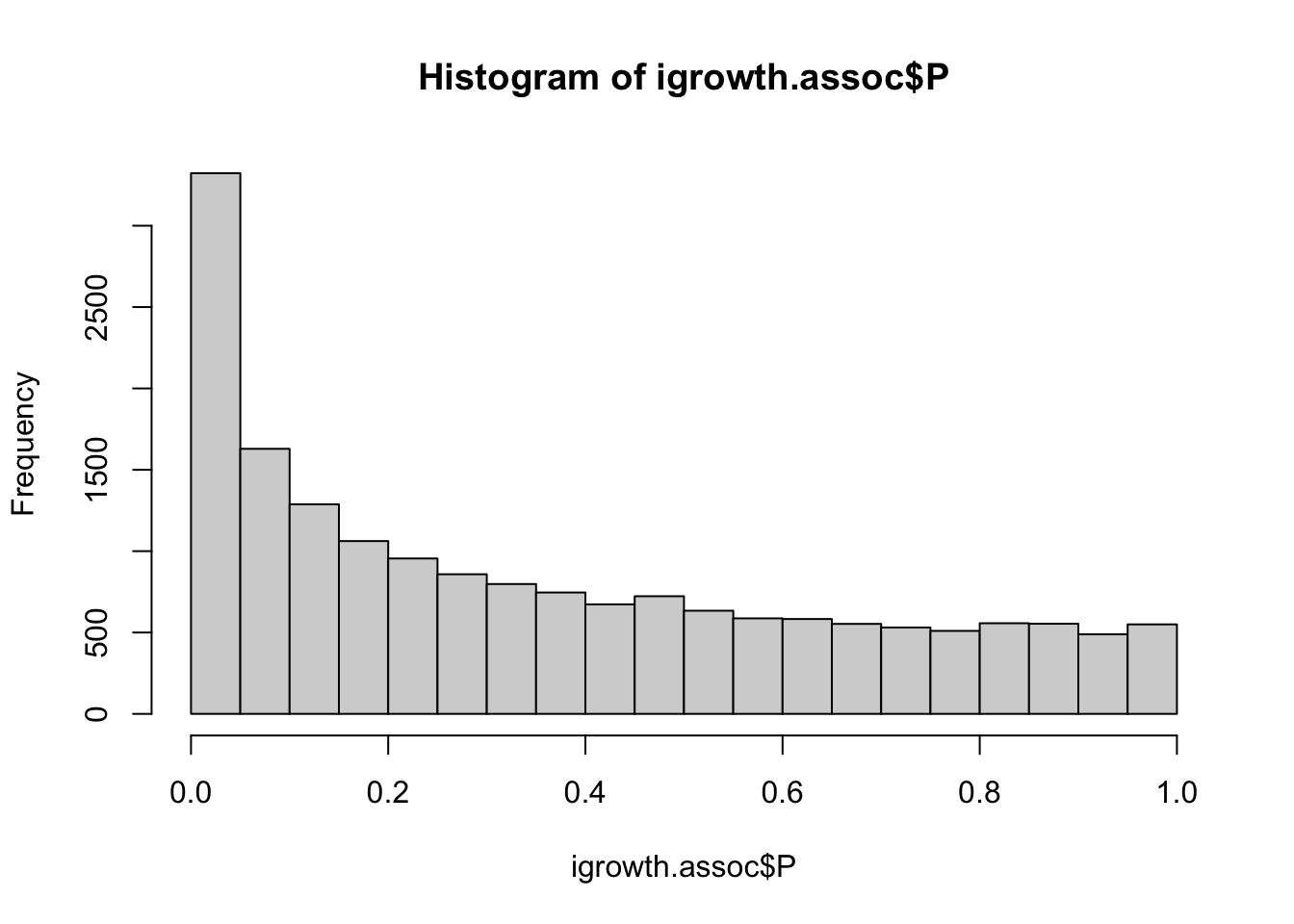
qqunif(igrowth.assoc$P)
## install.packages("qqman")
library(qqman)## ## For example usage please run: vignette('qqman')## ## Citation appreciated but not required:## Turner, S.D. qqman: an R package for visualizing GWAS results using Q-Q and manhattan plots. biorXiv DOI: 10.1101/005165 (2014).## manhattan(igrowth.assoc, chr="CHR", bp="BP", snp="SNP", p="P" )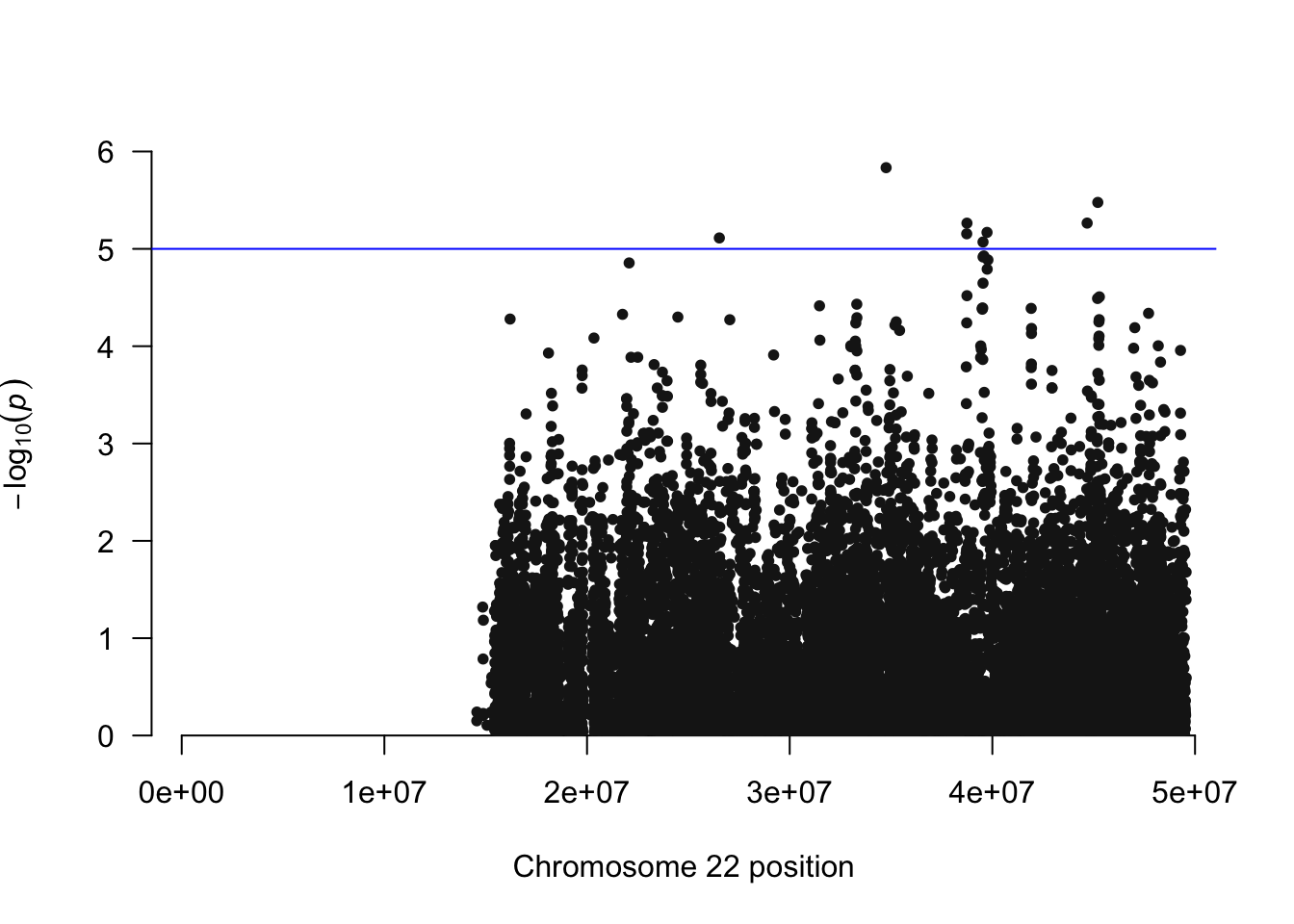
Calculate principal components using plink
## generate PCs using plink
if(!file.exists(glue::glue("{work.dir}output/pca.eigenvec")))
system(glue::glue("~/bin/plink --bfile {work.dir}hapmapch22 --pca --out {work.dir}output/pca"))
## read plink calculated PCs
pcplink = read.table(glue::glue("{work.dir}output/pca.eigenvec"),header=F, as.is=T)
names(pcplink) = c("FID","IID",paste0("PC", c(1:(ncol(pcplink)-2))) )
pcplink = popinfo %>% left_join(superpop,by=c("population"="Population")) %>% inner_join(pcplink, by=c("FID"="FID", "IID"="IID"))
## plot PC1 vs PC2
pcplink %>% ggplot(aes(PC1,PC2,col=population,shape=SuperPop)) + geom_point(size=3,alpha=.7) + theme_bw(base_size = 15)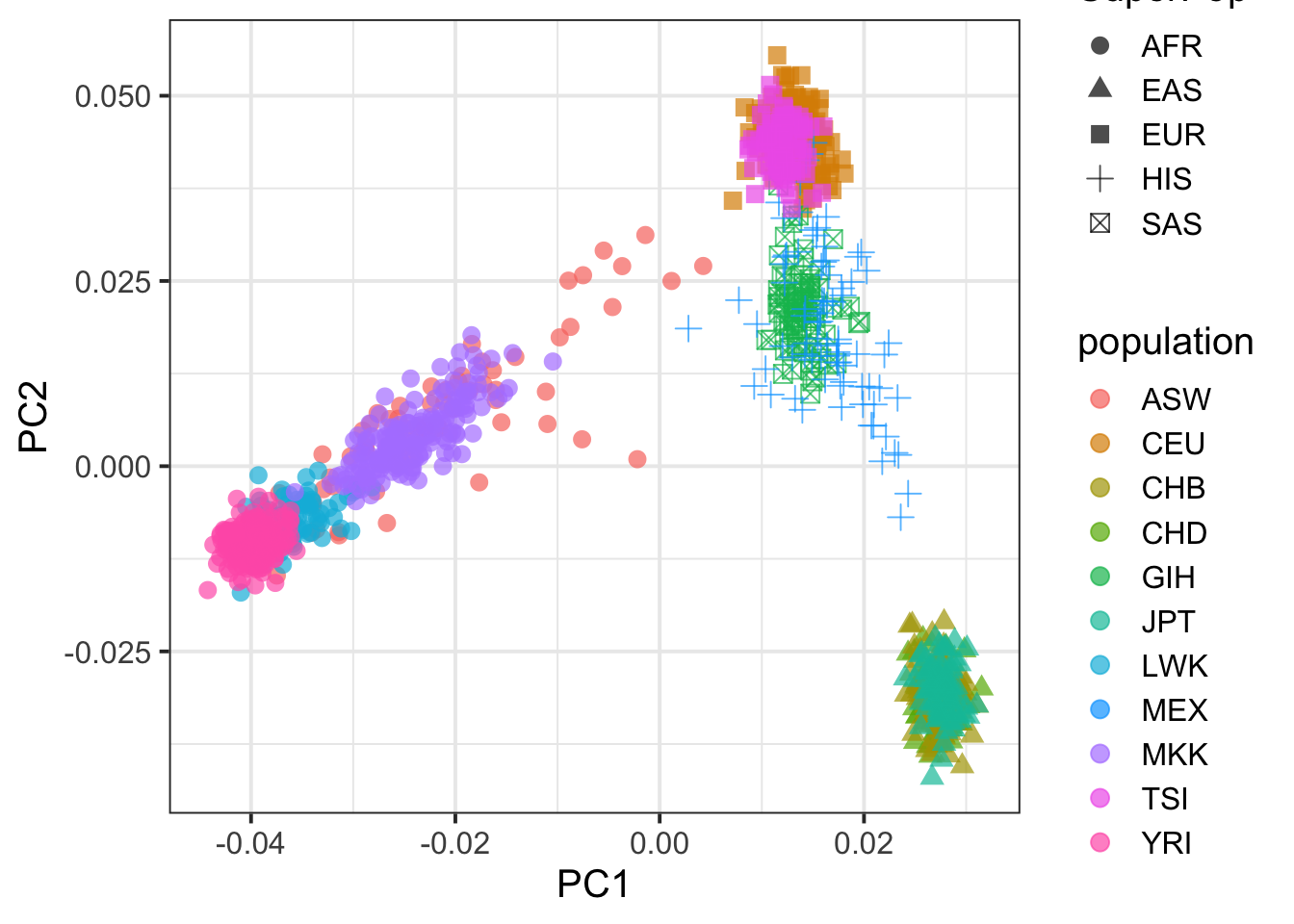
runnig igrowth GWAS using PCs
if(!file.exists(glue::glue("{work.dir}output/igrowth-adjPC.assoc.linear")))
system(glue::glue("~/bin/plink --bfile {work.dir}hapmapch22 --linear --pheno {work.dir}igrowth.pheno --pheno-name growth --covar {work.dir}output/pca.eigenvec --covar-number 1-4 --hide-covar --maf 0.05 --out {work.dir}output/igrowth-adjPC"))
igrowth.adjusted.assoc = read.table(glue::glue("{work.dir}output/igrowth-adjPC.assoc.linear"),header=T,as.is=T)
##indadd = igrowth.adjusted.assoc$TEST=="ADD"
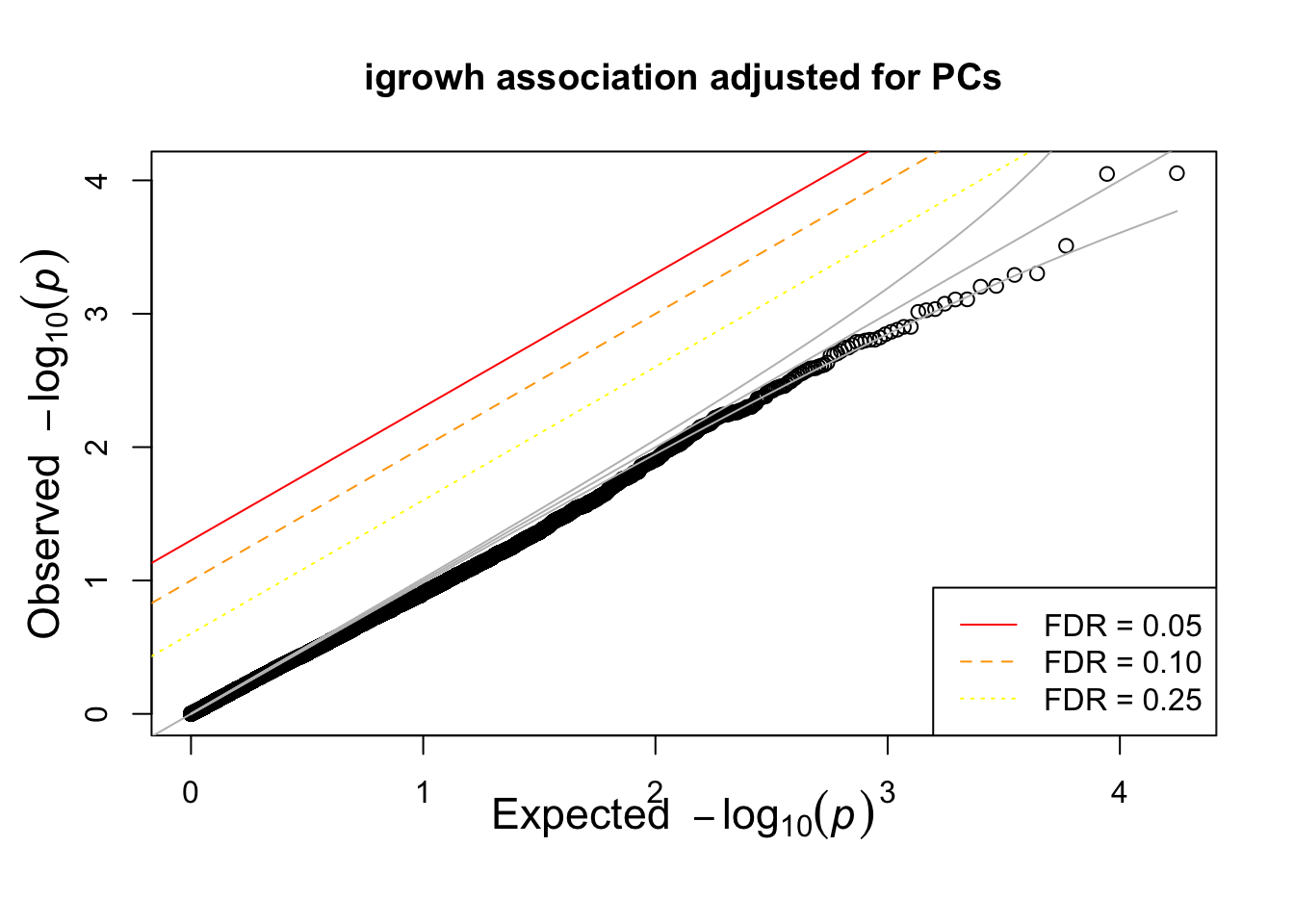
titulo = "igrowh association adjusted for PCs"
hist(igrowth.adjusted.assoc$P,main=titulo)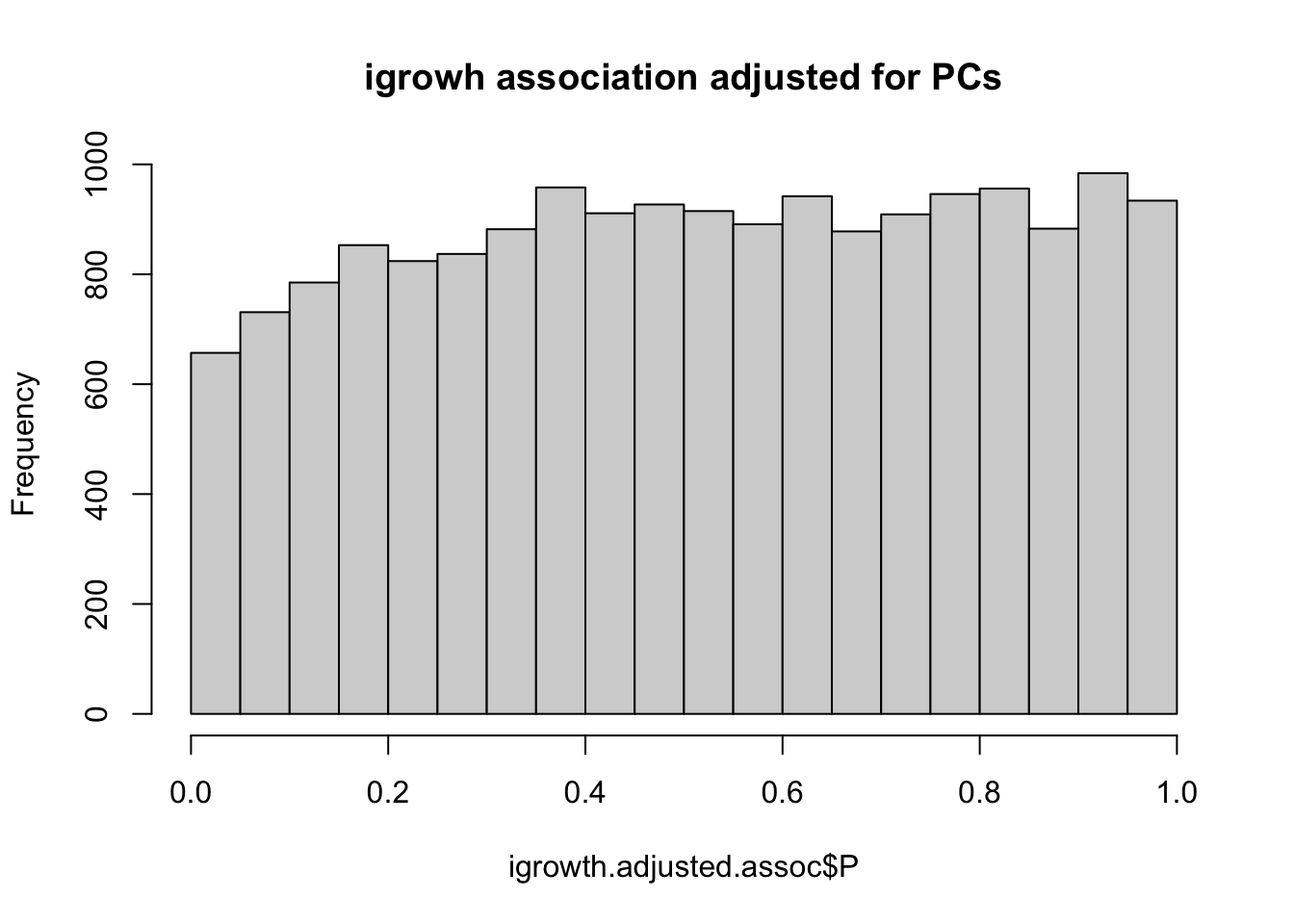
qqunif(igrowth.adjusted.assoc$P,main=titulo)